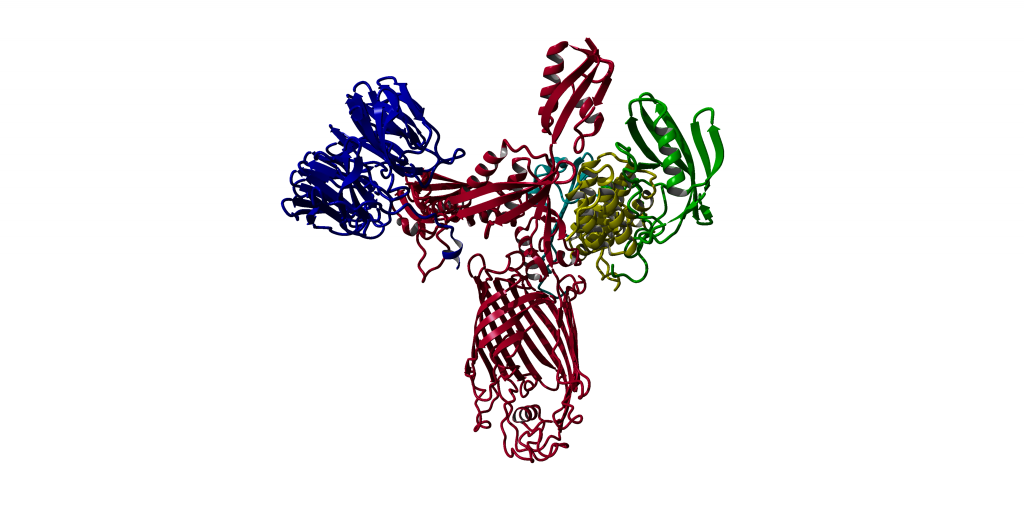
Molecular Simulations
Background
The overprescription and misuse of antibiotics over the past century has produced a growing crisis: multidrug-resistant (MDR) bacteria. Strains that were once trivially treatable now require last-resort drugs, and a meaningful fraction are becoming untreatable entirely. Developing novel antibiotics that evade existing resistance mechanisms is one of the most pressing problems in modern medicine.
Antimicrobial peptides (AMPs) — specifically bacteriocins produced by competing bacteria — represent a promising class of candidates. Unlike conventional antibiotics, they often target conserved structural machinery that bacteria cannot easily mutate away. This project focuses on one such target: the BAM complex, a conserved outer membrane protein assembly system present in gram-negative bacteria.
Objective
The goal is to explore docking interactions between bacteriocins and BamA, the central subunit of the BAM complex, across two closely related Bacteroides species: B. fragilis (clinically vulnerable) and B. vulgatus (resistant). Understanding the structural basis for this difference could guide the design of targeted antimicrobial agents.
This work was conducted under the mentorship of Thiago Monteiro Araujo dos Santos, PhD.
Progress
Homology models for both BamA variants were built using the YASARA twinset, starting from known crystal structures of related proteins. Working at roughly 25% sequence identity places this squarely in the "twilight zone" of homology modeling — structural predictions become less reliable and require more careful validation.
Model refinement was performed using YASARA and ModRef, with particular attention to the POTRA domain, which is the extracellular region most likely to interact with incoming bacteriocins. Model quality was assessed using Swiss-Model's Qmean scoring tool to evaluate local and global structural plausibility before moving to docking.

Next Steps
With validated homology models in hand, the next phase involves molecular dynamics simulation using membrane lipid environments to more accurately model the protein's native conformational behavior. AlphaFold predictions are also being evaluated as an independent validation reference.
Docking simulations will be run using Piper on the Schrödinger platform, accessed via the Northeastern University research computing cluster. The objective is to identify candidate bacteriocins that bind selectively to the vulnerable B. fragilis variant but not the resistant B. vulgatus — a structural selectivity profile that could serve as a starting point for antibiotic development.